


It has been over 50 years since the first proposals were put forward in the UK for non-medical healthcare professionals to expand their scope of responsibilities (Castledine, 2003). Although the specific nature of these responsibilities was uncertain at that time, what began with a few experimental posts eventually led to the emergence of advanced clinical practitioners (ACPs) in the early 2000s. These roles continued to evolve as a response to the growing need to expand the healthcare workforce, improving access to care, and fully using skills and expertise to meet changing societal needs. In the future, the UK has set an ambitious goal to train over 6000 ACPs annually, with particular emphasis on addressing the demand for specialised services (NHS England, 2023). Their growing presence across healthcare means that those living with rarer diseases will now more regularly be reviewed by ACPs, and these practitioners will increasingly need a solid grounding in the management principles of these conditions.
Interstitial lung disease (ILD) is a key example; it incorporates a group of over 300 diseases, which are characterised by the presence of inflammation and/or fibrosis within the lung parenchyma (Hilberg et al, 2022). This diversity is reflected in the range of underlying aetiologies, which include autoimmune disease, drugs and environmental exposures, although many have no clearly identifiable cause. ILDs can be lung-specific or associated with multi-organ disease and as the disease progresses, the impact on quality of life and daily functioning can become substantial. Therefore, management requires a multidisciplinary approach drawing on the expertise from different specialties and members of the healthcare team, with ACPs taking a key role.
ILD diagnoses have been rising over the past 20 years, with an estimated prevalence of approximately 200 per 100 000 people across Europe (Hilberg et al, 2022). These numbers are likely to increase as screening programmes for lung cancer are scaled up, alongside greater use of cross-sectional CT imaging for unrelated conditions. Recognition of ILDs in primary care remains relatively low (Antoniou et al, 2014) and, with ACPs regularly reviewing patients presenting with respiratory symptoms, increased awareness of ILDs will improve timely investigation and diagnosis. ILDs with a progressive fibrotic phenotype, such as idiopathic pulmonary fibrosis (IPF), are life-limiting and have a prognosis poorer than many cancers (Hilberg et al, 2022); therefore, earlier diagnosis and treatment initiation is likely to have significant patient benefit.
Once an ILD is suspected, referral to a respiratory centre with access to specialist multidisciplinary team (MDT) review is recommended to establish a diagnosis and facilitate further management. To improve diagnostic efficiency/patient outcomes, regional centres have established distinct referral routes spanning from primary to tertiary care; familiarity with a local pathway is required when considering a referral. The potential of treatment to influence disease trajectory is variable and partly depends on the balance between inflammatory and fibrotic components. Treatment can be largely divided into two broad categories; agents that target the (potentially reversible) inflammatory or ‘cellular’ disease, and agents that aim to slow development of fibrosis (Bradley et al, 2008).
A comprehensive assessment of how the disease impacts the individual should be integral to any management pathway. Patients can present with significant unmet needs that can adversely impact both respiratory symptoms and quality of life. Patient requirements and preferences are highly individual, will evolve throughout the disease course, and can have a profound impact on their families and caregivers. Supportive care includes non-pharmacological interventions, such as pulmonary rehabilitation, smoking cessation, occupational therapy and psychological support. Disease education and awareness promotes self-management and healthy living, which in turn can help patients identify early signs of deterioration. Delivery of care should be designed to meet the complex needs of these patients and close collaboration with the patient at all stages of their clinical course is essential. This article will cover the diagnostic, treatment and management principles of ILD.
Route to diagnosis
Practitioners across healthcare are all too aware that the classic respiratory symptoms of exertional dyspnoea and non-productive cough are in themselves non-specific. The multitude of ILDs, with individual complexities and lack of a single definitive diagnostic test, highlights the challenge facing those in primary and secondary care. Chest auscultation and spirometry can assist in differentiating between airways and interstitial disease; however, in a world where virtual review is increasingly used, there is a risk of missing tell-tale features on history alone, especially as symptoms can develop slowly over several years (Bokolo, 2020). Supporting examination features include fine inspiratory crackles (‘Velcro’ crackles) on chest auscultation, most noted at lung bases. Digital clubbing can also be a feature, although prevalence is variable depending on the ILD, with estimates falling between 7% and 42% (van Manen et al, 2016). Connective tissue disease (CTD) is a diverse group of inflammatory disorders to which the lung is particularly vulnerable. CTDs can co-exist or even present initially with an ILD, so the presence of CTD symptoms and/or signs such as Raynaud's, arthritis, arthralgia, myalgia and sicca symptoms should increase suspicion of an ILD (Wells and Denton, 2014).
Radiology is integral to the diagnosis and management of respiratory disease. While chest X-ray can be a useful screening test (Wielpütz et al, 2014), early ILD can be challenging to diagnose and X-rays may appear unremarkable (Thickett et al, 2014). High-resolution computed tomography (HRCT) allows for characterisation of lung tissue abnormalities through submillimetre cross-sectional images of the lung fields (Isozaki et al, 2023). However, this detailed characterisation cannot discriminate potential aetiology, and therefore a comprehensive respiratory assessment, including exposure history and identification of risk factors, is essential. Subsequent discussion and consensus opinion at a specialist ILD MDT has improved accuracy of diagnosis and reduced the requirement for biopsy, which is associated with significant morbidity (Raghu et al, 2022).
Radiology is not only a diagnostic tool but also valuable in assessing disease progression and treatment response. Repeat CT imaging can define or potentially redefine the diagnosis; while some initially ‘unclassifiable’ ILDs develop over time into a more characteristic pattern, others can evolve from one pattern to another. Predominantly inflammatory diseases can develop into a more fibrotic pattern; examples of this include connective tissue associated-ILD (CTD-ILD) and hypersensitivity pneumonitis (HP) (Bradley et al, 2008; Raghu et al, 2022). Identifying the relative proportion of the fibrotic and inflammatory component is important as it impacts the initial treatment strategy adopted (Figure 1).
Principles of management
Inflammatory predominant
ILD diseases can be inflammatory or fibrotic, but often have components of both. Treatment options for inflammatory-predominant ILD are similar to other autoimmune/inflammatory diseases and revolve around use of immunosuppressive agents (van den Bosch et al, 2022). Unless there is a contraindication, prednisolone remains first-line therapy for ILDs such as non-specific interstitial pneumonitis (NSIP), CTD-ILD, HP, sarcoidosis and organising pneumonia (Nunes et al, 2015; Raghu et al, 2022). Initial treatment doses are similar across all ILDs, at approximately 0.5mg/kg, with a weaning plan over a few months, followed by clinical review to determine symptomatic and physiological response. Cumulative dose side effects from long-term and intermittent use of steroids can result in significant morbidity. Adverse effects include osteoporosis, hypertension, diabetes and weight gain (Manson et al, 2009). Therefore, second-line agents are often introduced to minimise steroid burden if significant medium-to long-term immunosuppression is required.
Steroid-sparing agents include mycophenolate, azathioprine and methotrexate. While they lack the side effect profile of prednisolone, other adverse effects can occur, most commonly gastrointestinal disturbance, liver dysfunction and leucopoenia (van den Bosch et al, 2022). There is increased risk of benign and malignant tumours (most notably skin cancers) in those on long-term treatment and patients should be counselled regarding this. Increased susceptibility to infection is also a risk that needs to be considered, especially given that infection itself may drive progression of the ILD (Azadeh et al, 2017). The threshold for commencing treatment may depend on the underlying disease. More aggressive ILDs, such as pulmonary vasculitis (Alba et al, 2017) and anti-synthetase syndrome (Witt et al, 2016) may warrant therapeutic intervention even when the disease is mild because of the higher risk of disease progression. In other diseases that tend to follow a more indolent course, a watch-and-wait strategy may be advocated, looking for evidence of disease progression before treatment is initiated.
The aim of immunosuppressive treatment is to reduce inflammation and prevent (or at least reduce) the development of fibrosis. This follows the doctrine that chronic inflammation is a prerequisite to fibrosis, with inflammatory cells eventually promoting the deposition of scar tissue. However, more recent studies indicate that fibrosis can develop along different pathways, which perhaps explains the lack of apparent efficacy of immunosuppressive agents in fibrotic lung diseases such as IPF. As an ILD evolves, the balance can shift away from inflammation and towards fibrotic disease, resulting in progressive fibrosis despite immunosuppression (Renzoni et al, 2021).
Progressive fibrosis
Progressive fibrosing interstitial lung disease (PF-ILD) refers to any ILD where there is a progressive accumulation of fibrotic tissue. IPF is the archetypal example of this, although other ILDs can evolve over time in a similar manner, with CTD-ILD and HP being notable examples. The underlying mechanisms driving the progression of fibrosis remain incompletely understood, although a large body of research has been undertaken in the context of IPF to identify the fibrotic pathways driving the disease process. It is hypothesised that repetitive injury to the alveolar epithelial cells sets in motion a cascade of profibrotic mediators, which leads to the recruitment, proliferation and aberrant activation of collagen-secreting fibroblasts. The result is progressive accumulation of extracellular matrix that gradually replaces normal lung tissue (Moss et al, 2022), resulting in respiratory compromise and a poor prognosis, which has a median survival of only 3–5 years after an IPF diagnosis.
Advances in understanding of the disease have resulted in the development of drugs that specifically target fibrogenic pathways; two are currently licenced for this disease. Pirdenidone was approved in the UK in 2011 and is available to patients with lung function tests within defined criteria (National Institute for Health and Care Excellence (NICE), 2018). Its exact mechanism of action remains unknown, but studies have shown that it inhibits the production of profibrotic cytokines and reduces collagen deposition (Schaefer et al, 2011). Nintedanib, a triple kinase inhibitor that blocks downstream profibrogenic pathways through inhibition of cytokines, was the second drug approved for use in the UK (NICE, 2016) and has shown similar efficacy to pirfenidone in reducing fibrotic progression (Finnerty et al, 2021). Recognition that fibrotic pathways in other ILDs are likely to have significant overlap to IPF led to the INBUILD study. This evaluated the use of nintedanib in a range of progressive fibrotic ILDs and found similar outcomes to IPF (Flaherty et al, 2019). This has resulted in the licensing of this agent in PF-ILD in the UK, where there is evidence of disease progression (NICE, 2021).
Patient–clinician partnership
Any treatment decision should be viewed as a partnership between the clinician and the patient. Discussion of treatment should be realistic and emphasise the aims from the outset (often to slow, rather than stop or reverse the disease). Patients should be aware that side effects may negatively impact on quality of life. Clinical decisions must fit with the patient's values, and expectations must be set appropriately. Educational resources tailored to help patients make informed decisions and support engagement are helpful. Self-management interventions have been more extensively studied in airways disease, such as chronic obstructive pulmonary disease (Baker and Fatoye, 2019), where interventions involving diet, exercise and cognitive behavioural therapy have been shown to improve hospital admissions and levels of dyspnoea. A recent Delphi study emphasised the importance of individualisation, goal setting and feedback when considering self-management interventions in PF-ILD (Lee et al, 2022). Althobiani et al (2024) highlight that digital methods will play a crucial role in any comprehensive supported self-management programme; however, the development of customisable care pathways remains a work in progress.
Monitoring
Radiology
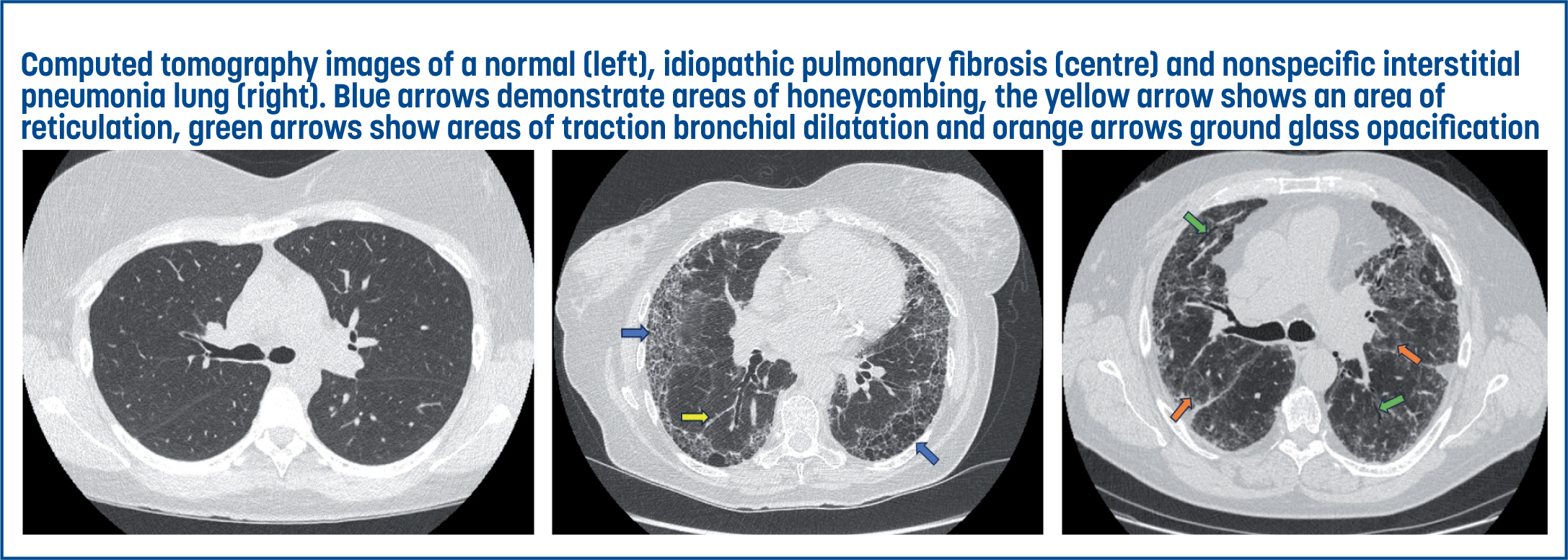
Regular monitoring is essential to evaluate disease trajectory in those living with ILD, and inform therapeutic decisions and patient counselling. The radiological appearance of inflammation on chest CT is that of increased opacification of the lung tissue, referred to as ‘ground-glass’ or consolidation, depending on the density of the abnormality (Hansell et al, 2008). Improvement of these radiological components indicates a positive response to treatment and the HRCT is important in this evaluation. In contrast, pulmonary fibrosis typically manifests as a distinctive pattern characterised by irregular, reticular opacities that appear as a network of lines, reflecting the deposition of fibrous tissue in the lung parenchyma. While other processes, such as pulmonary oedema, can cause reticulation, in fibrosis, this often appears in a subpleural and bibasal distribution, and may be accompanied by traction bronchial dilatation. Honeycombing represents the end stage of this process and is characterised by clustered cystic airspaces with well-defined walls (Hata et al, 2021). In the context of fibrosis, limiting further progression is considered a positive treatment response.
Lung function test
Lung function tests involve spirometry and gas transfer measures, and these provide evidence of a physiological response. Routinely available in secondary care and without radiation exposure concerns, they form an essential serial monitoring tool (Nathan et al, 2020). Forced vital capacity (FVC) is a simple spirometry measure of the maximum volume of air a person can exhale forcefully. The FVC value typically falls as the disease progresses, with drops greater than 10% being clinically significant; however, in early stages of disease or in those with a more inflammatory pattern, FVC may not always correlate as well with disease extent. Gas transfer is a more challenging lung function measure, and involves the patient inhaling carbon monoxide and holding their breath for around 10 seconds. This small molecule should easily diffuse across the alveolar membrane, but abnormalities of the lung tissue or surrounding blood vessels will reduce diffusion. As the patient breathes out, the residual carbon monoxide not absorbed is measured, therefore providing an indication of the integrity of the alveolar membrane and pulmonary vasculature (Doyle et al, 2012). A gas transfer change of more than 15% is considered significant but is not specific to ILD; other pathologies can cause a fall in value, such as the presence of pulmonary emboli or congestive cardiac failure. There is increasing real-world data that use of home spirometry could potentially support virtual review and early identification of acute decline in particular patient cohorts (Wijsenbeek et al, 2023); however, there remains a paucity of evidence on how to integrate this technology to promote greater patient self-management.
The 6-minute walk test and oxygen assessments
The 6-minute walk test (6MWT) is a simple investigation that can be undertaken by the ACP, and is valuable at initial assessment and follow-up of ILD patients (Rizzi et al, 2015). The trajectory of change has been shown to mirror FVC decline in IPF (Brown and Nathan, 2018), although confounding factors, such as mobility, need to be taken into consideration. In providing a functional measure of a patient's overall cardiopulmonary reserve, a 6MWT provides a distinct and valuable input to assessment.
Pulse oximetry is a key component of a 6MWT, both during exertion and to assess recovery time. Resting oxygen saturations should be above 92% and, unlike in chronic obstructive pulmonary disease, there is rarely a concern regarding hypoxic drive and the associated requirement to reduce target saturations. Indeed, hypercapnia is rarely seen outside of context of end-stage disease and is a poor prognostic indicator. Significant exertional desaturation below 88% is widely accepted as an indication for an ambulatory oxygen therapy (AMBOT) evaluation. There is evidence that AMBOT can increase exercise capacity by correcting exertional desaturation and this in turn can facilitate improvement in overall conditioning; however, many patients will experience practical issues as a result of the limitations of current ambulatory oxygen delivery devices, and the psychological impact of oxygen therapy should not be underestimated (Bell et al, 2017). Long-term oxygen therapy (for at least 15 hours a day) should be offered for those in respiratory failure.
Acute exacerbations
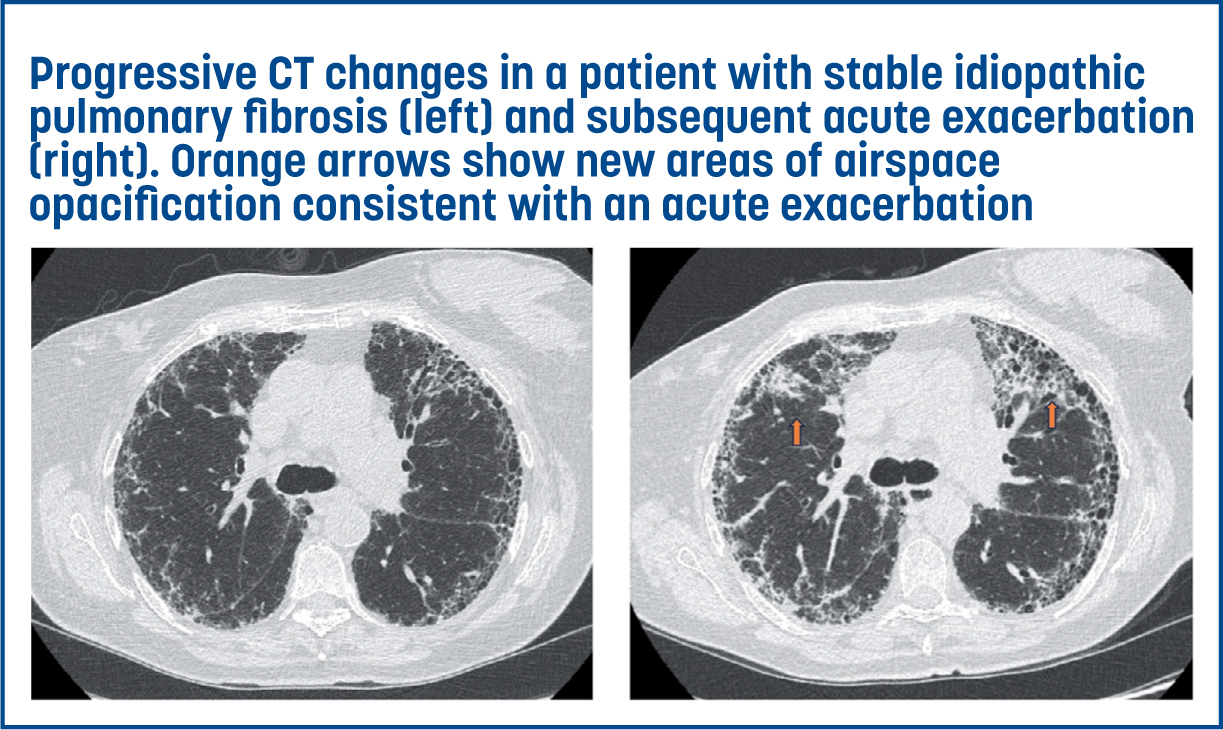
Acute exacerbation (AE) is a significant risk for those living with a PF-ILD. It is defined as a rapid worsening in dyspnoea (typically within 30 days or fewer), in association with new ground glass or multi-focal consolidative changes on CT (Collard et al, 2016) (Figure 2). Patients at greatest risk are those with IPF, where an annual incidence of 5–19% is reported (Kolb et al, 2018). The mechanisms driving these events have not been fully elucidated but there is evidence that these may represent superimposed inflammatory change or an acceleration of the underlying disease process (Johannson and Collard, 2013). Alternative causes of symptomatic decline should be excluded, such as heart failure, pneumothorax and pulmonary embolism. The prognosis of an AE is poor, with approximately a 50% rate of in-hospital mortality and a 90% rate of 6-month mortality. Pragmatically, management of AE events revolves around treatment of reversible causes, with prompt initiation of antibiotics if there is any evidence of infection. The recommendation for the use of steroids is based on consensus opinion, with the rationale that they may target an additional inflammatory component, although the clinical response is variable (Juarez et al, 2015). The risk of sudden irreversible deterioration from AE makes early discussion regarding the clinical course and prognosis important and advanced care planning vital.
Symptom management
Given the significant morbidity associated with ILD (alongside other chronic respiratory disease), an important goal of management is to maintain and support quality of life. There are several major pharmacological and non-pharmacological interventions that can be conducted to support symptom management in ILD (Table 1).
| Symptomatic concern | Treatment or intervention |
|---|---|
| Dyspnoea (Ryerson et al, 2012) | Pulmonary rehabilitation |
| Cough (van Manen et al, 2016) | Cough management |
| Anxiety/depression (Fulton and Ryerson, 2015; Pescheny et al, 2020) | Referral to talking therapy |
| Weight loss or gain (Fulton and Ryerson, 2015) | Pulmonary rehabilitation |
| Co-morbidities (Fulton and Ryerson, 2015) | Evaluation of pulmonary hypertension via specialist review |
Breathlessness
Dyspnoea is the major symptom and is often multifactorial, contributed to by co-morbidities and psychological factors (Banzett and O'Donnell, 2014). Fear of breathlessness by patients and their loved ones can lead to reduced activity as a strategy to manage symptoms, which then accelerates deconditioning. Referral to pulmonary rehabilitation programmes is recommended and has demonstrated improved dyspnoea, physical activity and quality-of-life measures (Jastrzebski et al, 2006). Non-pharmacological approaches such as breathing techniques and fan therapy are also important management tools and should be initiated early. The anxiety-breathlessness cycle is well documented and techniques such as cognitive behavioural therapy and mindfulness can be powerful tools (Spathis et al, 2017). Such symptom management support is being redefined through increasing use of video-conferencing and mobile applications, complemented by the use of wearables and remote monitoring devices. This facilitates a more tailored and accessible delivery of interventions for those living with ILD, including pulmonary rehabilitation and behavioural therapies (Holland et al, 2021).
As breathlessness progresses, introduction of a low-dose opioid may be considered. While the suggestion of opioids can be negatively emotive for some, morphine at doses of 2.5mg up to four times daily can be a valuable adjunct in relieving breathlessness and is unlikely to cause significant side effects such as drowsiness, constipation or dependence. Clear guidance should be given to focus use around anticipated periods of exertion, such as morning and night-time routines. For those with advanced disease and severe dyspnoea, long-acting morphine can be beneficial. Where anxiety is a clear driver, low-dose lorazepam (0.5mg three times daily) can be helpful, or mirtazapine for those with more chronic anxiety (Morélot-Panzini, 2017). Integration of palliative care into routine support for those living with PF-ILD is strongly recommended, with active disease management and palliation of symptoms viewed as complementary (Janssen et al, 2023). Delivery of personalised care for individuals often with substantial comorbidities, necessitates close collaboration and effective communication between specialised ILD services and community teams.
Cough
Cough can be a debilitating symptom in ILD. It is usually dry, with a high sputum load being an uncommon feature (van Manen et al, 2016). For those with features of bronchiectasis, sputum clearance techniques can be of value, alongside mucolytics such as carbocisteine. Ensuring adequate control of reflux can also improve symptoms. Non-pharmacological options for those with refractory chronic cough include education, laryngeal hygiene, adequate hydration and cough suppression techniques. Pharmacological treatment for chronic cough includes opioids, such as codeine linctus or modified release morphine (Wu et al, 2022), and neuropathic agents, such as gabapentin (although side effects may be limiting) (Song et al, 2020). There is also some evidence that tiotropium can have antitussive effect in those with fibrotic disease (Birrell et al, 2014). Empirical trials of treatment are often necessary, with no single intervention completely alleviating symptoms.
Non-respiratory
Risk of malnutrition increases with disease progression and can be exacerbated by medical treatments. This can result in a negative cycle impacting exercise capacity, quality of life and mortality (Rinaldi et al, 2017). Access to educational materials on nutrition supported by dietitian review can impact positively on patient outcomes. Frailty is an additional risk factor associated with poor prognosis for those with PF-ILD and needs to be taken into account when considering treatment strategies (Rinaldi et al, 2017). Indeed, side effects from medications prescribed in ILD, particularly regarding anti-fibrotic agents, are often more pronounced and problematic in older, frailer individuals may be avoided for this reason. A validated frailty score can be helpful in assisting clinical decision making (Guler et al, 2020).
As the disease progresses over time, symptoms become more problematic and impact everyday tasks. This can precipitate low mood, with depression more commonly reported in those living with an ILD. Although routine screening for depression is rarely carried out during clinical ILD consultations, when this aspect was examined using a validated tool, depression was identified in 23% of ILD patients, and in 7%, it was considered clinically significant (Akhtar et al, 2013). Predictors for depression include severity of symptoms, poor functional status, reduced sleep quality, pain and low spirometry values. In those where steroid therapy has been initiated, there is also an associated risk of mood disturbance with potential for psychosis. Depression is also associated with poor treatment concordance (DiMatteo et al, 2000). Screening those with chronic illness causing functional disability (such as in PF-ILD) may be beneficial, and several tools are available for this indication (Akhtar et al, 2013).
Complications associated with progressive interstitial lung disease
Gastro-oesophageal reflux (GOR) as a driver for pulmonary fibrosis has long been proposed and has been most extensively studied in IPF (Kreuter and Raghu, 2018). GOR is common in IPF, and silent aspiration has been proposed to play a pathogenic role in mediating disease progression through recurrent injury to the alveolar epithelium (Kreuter and Raghu, 2018). Furthermore, progressive fibrosis may itself exacerbate reflux by increasing negative intrathoracic pressure and distorting mediastinal structures as the lung tissue contracts. Small studies have shown potential benefit of a proton pump inhibitor; however, the evidence is poor and routine antacid treatment in absence of symptoms is not currently recommended in IPF (Raghu et al, 2022). A multi-centre trial of use of lansoprazole in those with IPF in currently underway to evaluate this further (Treating People with Idiopathic Pulmonary Fibrosis with the Addition of Lanzoprazole, 2022). Fundoplication has also been examined as an anti-reflux measure for specific groups of patients with severe lung disease (such as those awaiting transplant) to reduce morbidity and stabilise disease; however, in their systematic review, Khor et al (2022) identified no statistically significant benefit.
Pulmonary hypertension (PH) is a recognised complication of ILD and is present in up to 30% of patients (Lettieri et al, 2006). PH exacerbates dyspnoea and impacts negatively on prognosis. In the absence of clinical features of PH, supportive features include a disproportionate reduction in gas transfer compared with FVC and dilatation of the pulmonary artery on CT imaging. Biomarkers such as B-type natriuretic peptide may be raised, but not always (Nathan et al, 2019). Echocardiogram is the most common screening tool, which often provides an estimate of probability. Right-heart catheterisation is required for definitive diagnosis, although consideration must be given as to whether a confirmed result would have therapeutic implications. Indeed, standard management of PH in the UK currently comprises of optimising the treatment of the underlying ILD, use of diuretics and long-term oxygen therapy, if clinically indicated (Nathan et al, 2019).
There also appears to be an increased risk of venous thromboembolic disease in certain ILDs. In IPF, a possible explanation is that the microenvironment is procoagulant and contributed by the reduction in mobility as a result of dyspnoea and co-morbidity (Margaritopoulos et al, 2017). It has been estimated that the risk of pulmonary embolism for those with sarcoidosis is approximately double that of the general population, although the mechanism for this is unclear (Swigris et al, 2011). Computed tomography pulmonary angiography is the standard investigation to identify thromboembolism; this imaging also provides useful ancillary information, such as evidence of radiological progression of ILD or evidence of heart failure. V/Q scanning is not recommended in ILD, because of the abnormal lung parenchyma (Young and Brown, 2011).
Conclusions
ILD encompasses a wide spectrum of diseases; ACPs across healthcare settings will increasingly review patients at various stages of their disease and treatment pathway. The principles of management need to consider not only the disease, but the trajectory and prognosis with and without treatment. Most importantly, the views and health beliefs of the patient must take centre stage, with careful assessment and counselling prior to commencing treatment required. It is important to recognise that multimorbidity is common and may impact both symptoms and treatment tolerance. Considering the holistic needs of the patient is essential and a focus on quality of life should be a priority, particularly in older and more frail patients.
ACPs play a significant role in supporting those living with chronic and often life-limiting disease. In diseases such as ILD, they can enable timely access of specialised expertise and co-ordinate multidisciplinary care. ACPs are also well placed to make holistic assessments, discuss treatment options, enhance patient education and provide self-management strategies. This comprehensive approach enhances ILD management and improves patient outcomes.